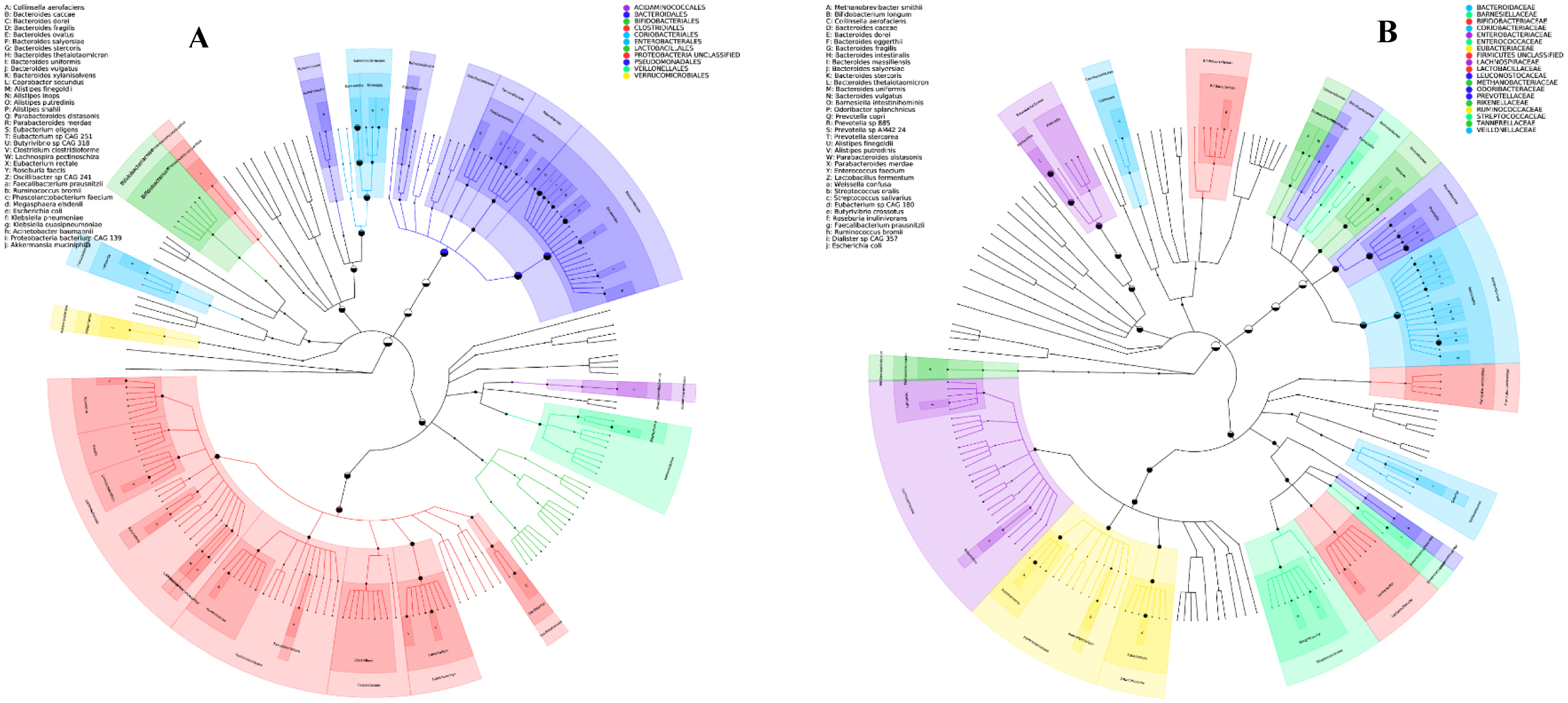
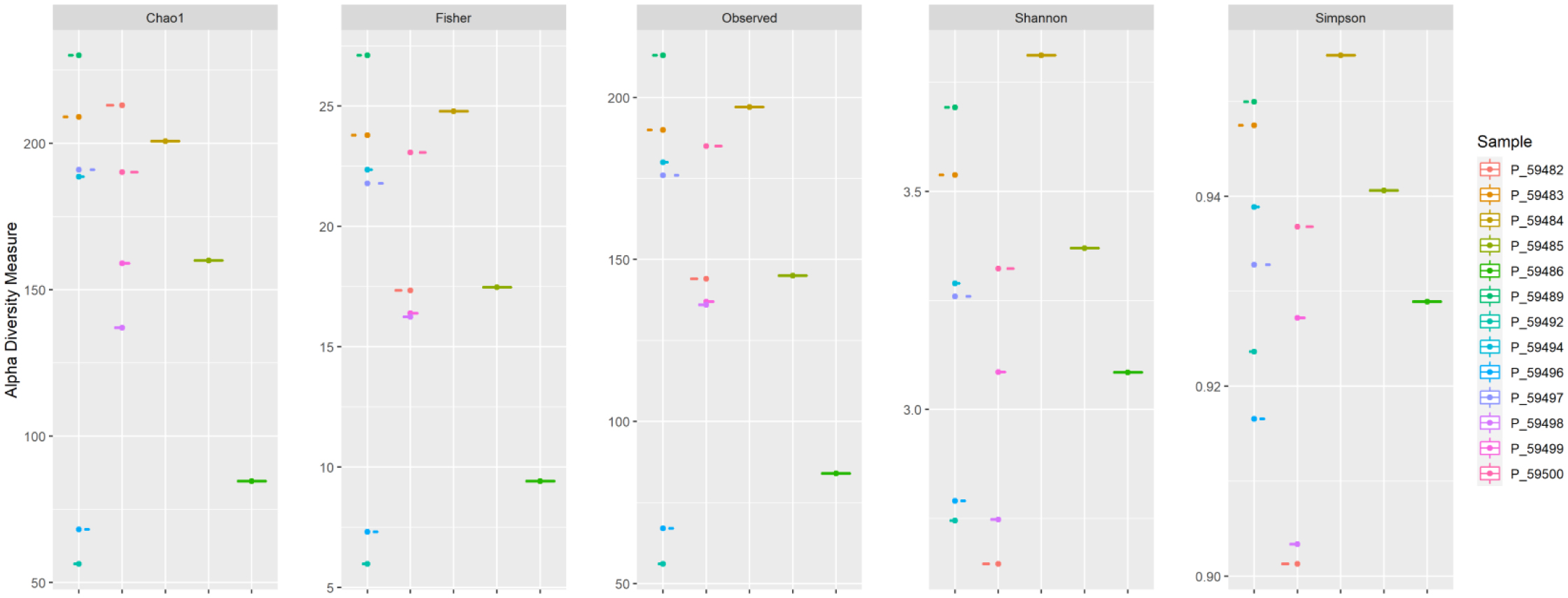
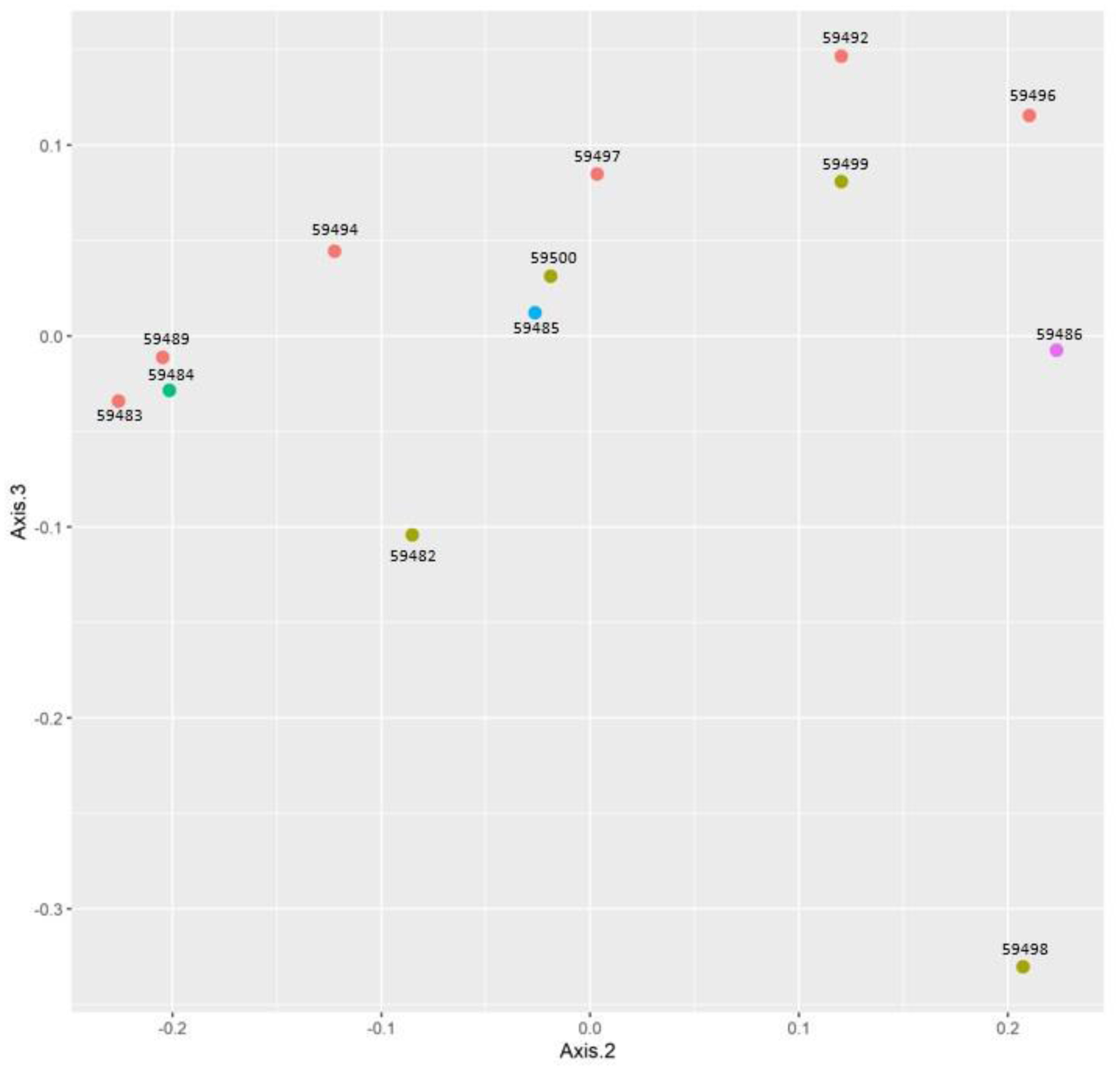
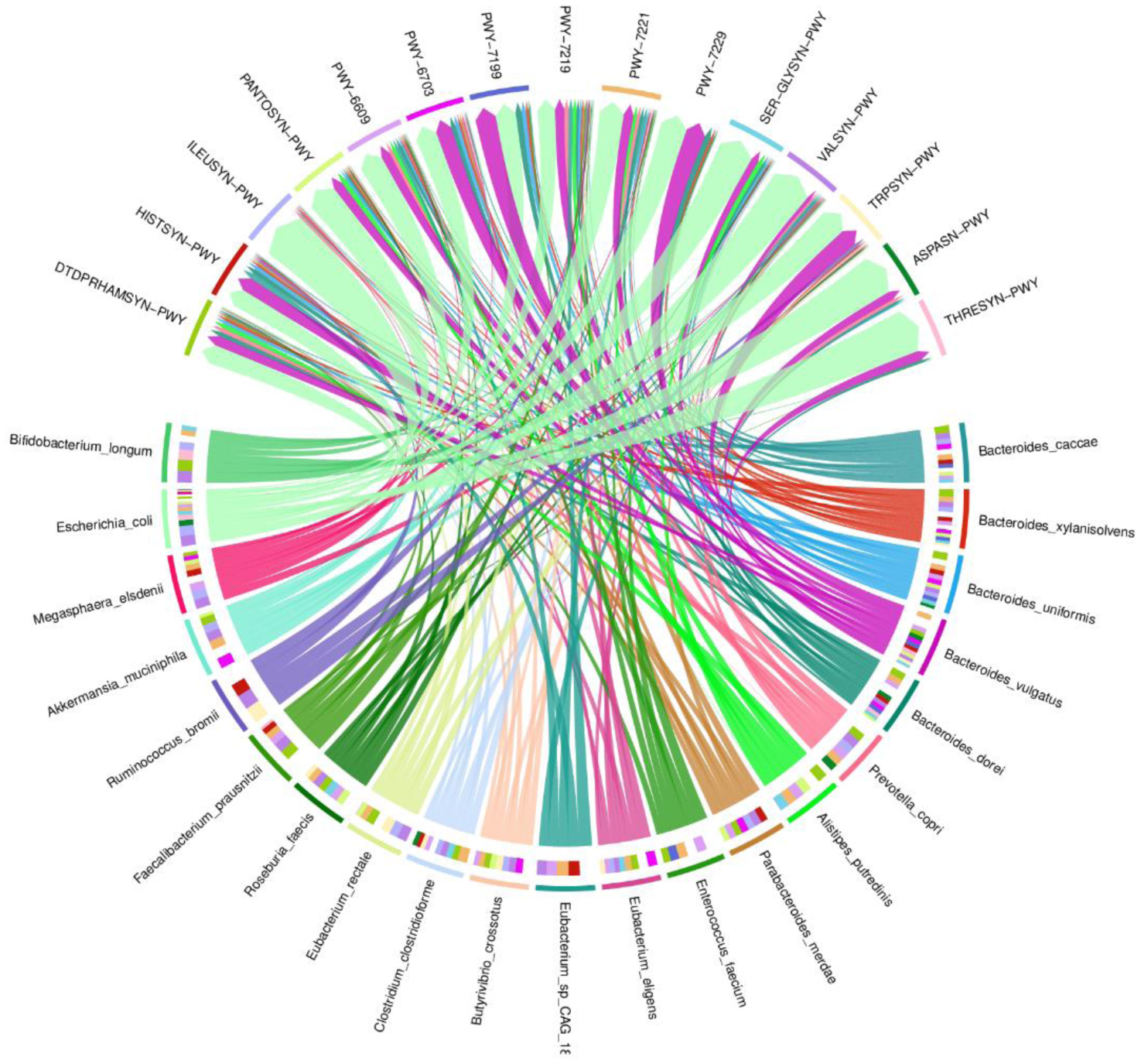
One of the most prevalent malignancies that significantly affects world health is colorectal cancer (CRC). While genetics are involved in a portion of CRC patients, most cases are sporadic. The microbiome composition could be a new source of tumor initiation and progression. This research was conducted to investigate the microbiota composition of CRC patients post colectomy at taxonomic and functional levels. Using a next-generation sequencing approach, using an Illumina Novaseq 6000, the fecal samples of 13 patients were analyzed and the obtained data was subjected to a bioinformatics analysis. The bacterial abundance and uniqueness varied in CRC patients alongside differences in bacterial counts between patients. Bacteroides fragilis, Bacteroides vulgatus, Escherichia coli, and Fusobacterium nucleatum were among the pro-cancerous microorganisms found. Concurrently, bacteria linked to CRC progression were detected that have been previously linked to metastasis and recurrence. At the same time, probiotic bacteria such as Bifidobacterium dentium, Bifidobacterium bifidum, and Akkermansia muciniphila increased in abundance after colectomies. Additionally, numerous pathways were deferentially enriched in CRC, which emerged from functional pathways based on bacterial shotgun data. CRC-specific microbiome signatures include an altered bacterial composition. Our research showed that microbial biomarkers could be more usefully employed to explore the link between gut microbiota and CRC using metagenomic techniques in the diagnosis, prognosis, and remission of CRC, thereby opening new avenues for CRC treatment.
Citation: Rana H. Abo-Hammam, Mohammed Salah, Sarah Shabayek, Amro Hanora, Samira Zakeer, Randa H. Khattab. Metagenomic analysis of fecal samples in colorectal cancer Egyptians patients post colectomy: A pilot study[J]. AIMS Microbiology, 2024, 10(1): 148-160. doi: 10.3934/microbiol.2024008
One of the most prevalent malignancies that significantly affects world health is colorectal cancer (CRC). While genetics are involved in a portion of CRC patients, most cases are sporadic. The microbiome composition could be a new source of tumor initiation and progression. This research was conducted to investigate the microbiota composition of CRC patients post colectomy at taxonomic and functional levels. Using a next-generation sequencing approach, using an Illumina Novaseq 6000, the fecal samples of 13 patients were analyzed and the obtained data was subjected to a bioinformatics analysis. The bacterial abundance and uniqueness varied in CRC patients alongside differences in bacterial counts between patients. Bacteroides fragilis, Bacteroides vulgatus, Escherichia coli, and Fusobacterium nucleatum were among the pro-cancerous microorganisms found. Concurrently, bacteria linked to CRC progression were detected that have been previously linked to metastasis and recurrence. At the same time, probiotic bacteria such as Bifidobacterium dentium, Bifidobacterium bifidum, and Akkermansia muciniphila increased in abundance after colectomies. Additionally, numerous pathways were deferentially enriched in CRC, which emerged from functional pathways based on bacterial shotgun data. CRC-specific microbiome signatures include an altered bacterial composition. Our research showed that microbial biomarkers could be more usefully employed to explore the link between gut microbiota and CRC using metagenomic techniques in the diagnosis, prognosis, and remission of CRC, thereby opening new avenues for CRC treatment.

| [1] |
Guyton KZ, Loomis D, Grosse Y, et al. (2015) Carcinogenicity of tetrachlorvinphos, parathion, malathion, diazinon, and glyphosate. Lancet Oncol 16: 490-491. https://doi.org/10.1016/S1470-2045(15)70134-8 
|
| [2] | Suliman MA, Zamzam ML, Omar AT, et al. (2020) Clinicopathological profile of colorectal cancer patients in Suez Canal University Hospitals-Egypt. J Cancer Biol Res 8: 1127. |
| [3] |
Liu R, Hong J, Xu X, et al. (2017) Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat Med 23: 859-868. https://doi.org/10.1038/nm.4358
|
| [4] |
Zitvogel L, Daillère R, Roberti MP, et al. (2017) Anticancer effects of the microbiome and its products. Nat Rev Microbiol 15: 465-478. https://doi.org/10.1038/nrmicro.2017.44
|
| [5] |
Sun J, Chen F, Wu G (2023) Potential effects of gut microbiota on host cancers: focus on immunity, DNA damage, cellular pathways, and anticancer therapy. ISME J 17: 1535-1551. https://doi.org/10.1038/s41396-023-01483-0
|
| [6] |
Arthur JC, Perez-Chanona E, Mühlbauer M, et al. (2012) Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338: 120-123. https://doi.org/10.1126/science.1224820
|
| [7] |
Dekker E, Tanis PJ, Vleugels JLA, et al. (2019) Colorectal cancer. Lancet 394: 1467-1480. https://doi.org/10.1016/S0140-6736(19)32319-0
|
| [8] |
Bharti R, Grimm DG (2021) Current challenges and best-practice protocols for microbiome analysis. Brief Bioinform 22: 178-193. https://doi.org/10.1093/bib/bbz155
|
| [9] |
Olovo CV, Huang X, Zheng X, et al. (2021) Faecal microbial biomarkers in early diagnosis of colorectal cancer. J Cell Mol Med 25: 10783-10797. https://doi.org/10.1111/jcmm.17010
|
| [10] |
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114-2120. https://doi.org/10.1093/bioinformatics/btu170
|
| [11] |
Beghini F, McIver LJ, Blanco-Míguez A, et al. (2021) Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife 10: e65088. https://doi.org/10.7554/eLife.65088
|
| [12] |
Asnicar F, Weingart G, Tickle TL, et al. (2015) Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 3: e1029. https://doi.org/10.7717/peerj.1029
|
| [13] |
Abubucker S, Segata N, Goll J, et al. (2012) Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol 8: e1002358. https://doi.org/10.1371/journal.pcbi.1002358
|
| [14] |
Wieczorska K, Stolarek M, Stec R (2020) The role of the gut microbiome in colorectal cancer: where are we? where are we going?. Clin Colorectal Cancer 19: 5-12. https://doi.org/10.1016/j.clcc.2019.07.006
|
| [15] | Zakharzhevskaya NB, Tsvetkov VB, Vanyushkina AA, et al. (2017) Interaction of bacteroides fragilis toxin with outer membrane vesicles reveals new mechanism of its secretion and delivery. Front Cell Infect Microbiol 7: 308. https://doi.org/10.3389/fcimb.2017.00308 |
| [16] |
Kostic AD, Chun E, Robertson L, et al. (2013) Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 14: 207-215. https://doi.org/10.1016/j.chom.2013.07.007
|
| [17] |
Sánchez-Alcoholado L, Ordóñez R, Otero A, et al. (2020) Gut microbiota-mediated inflammation and gut permeability in patients with obesity and colorectal cancer. Int J Mol Sci 21: 6782. https://doi.org/10.3390/ijms21186782
|
| [18] |
Wang X, Jia Y, Wen L, et al. (2022) Porphyromonas gingivalis promotes colorectal carcinoma by activating the hematopoietic NLRP3 inflammasome. Cancer Res 82: 2196. https://doi.org/10.1158/0008-5472.CAN-22-1136
|
| [19] |
Huycke MM, Abrams V, Moore DR (2002) Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis 23: 529-536. https://doi.org/10.1093/carcin/23.3.529
|
| [20] |
Hou X, Zhang P, Du H, et al. (2021) Akkermansia muciniphila potentiates the antitumor efficacy of FOLFOX in colon cancer. Front Pharmacol 12: 725583. https://doi.org/10.3389/fphar.2021.725583
|
| [21] |
Roh YH, Park KJ, Byun KD, et al. (2019) Abdominal actinomycosis misconceived as intestinal lymphoma: Report of a case. Int J Surg Case Rep 60: 171-174. https://doi.org/10.1016/j.ijscr.2019.05.063
|
| [22] |
Garceau R, Bourque C, Thibault L, et al. (2016) First report of clostridium lavalense isolated in human blood cultures. Can J Infect Dis Med Microbiol 2016: 7231805. https://doi.org/10.1155/2016/7231805
|
| [23] |
Gupta A, Dhakan DB, Maji A, et al. (2019) Association of Flavonifractor plautii, a flavonoid-degrading bacterium, with the gut microbiome of colorectal cancer patients in India. MSystems 4: e00438-19. https://doi.org/10.1128/mSystems.00438-19
|
| [24] |
Wang W, Cui J, Ma H, et al. (2021) Targeting pyrimidine metabolism in the era of precision cancer medicine. Front Oncol 11: 684961. https://doi.org/10.3389/fonc.2021.684961
|
| [25] |
Thomas AM, Manghi P, Asnicar F, et al. (2019) Author Correction: Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat Med 25: 1948. https://doi.org/10.1038/s41591-019-0663-4
|
| [26] |
Ma Y, Zhang Y, Xiang J, et al. (2021) Metagenome analysis of intestinal bacteria in healthy people, patients with inflammatory bowel disease and colorectal cancer. Front Cell Infect Microbiol 11: 599734. https://doi.org/10.3389/fcimb.2021.599734
|
 microbiol-10-01-008-s001.pdf microbiol-10-01-008-s001.pdf |
 |
Figures(4) / Tables(1)

Rana H. Abo-Hammam, Mohammed Salah, Sarah Shabayek, Amro Hanora, Samira Zakeer, Randa H. Khattab. Metagenomic analysis of fecal samples in colorectal cancer Egyptians patients post colectomy: A pilot study[J]. AIMS Microbiology, 2024, 10(1): 148-160. doi: 10.3934/microbiol.2024008







 DownLoad:
DownLoad: