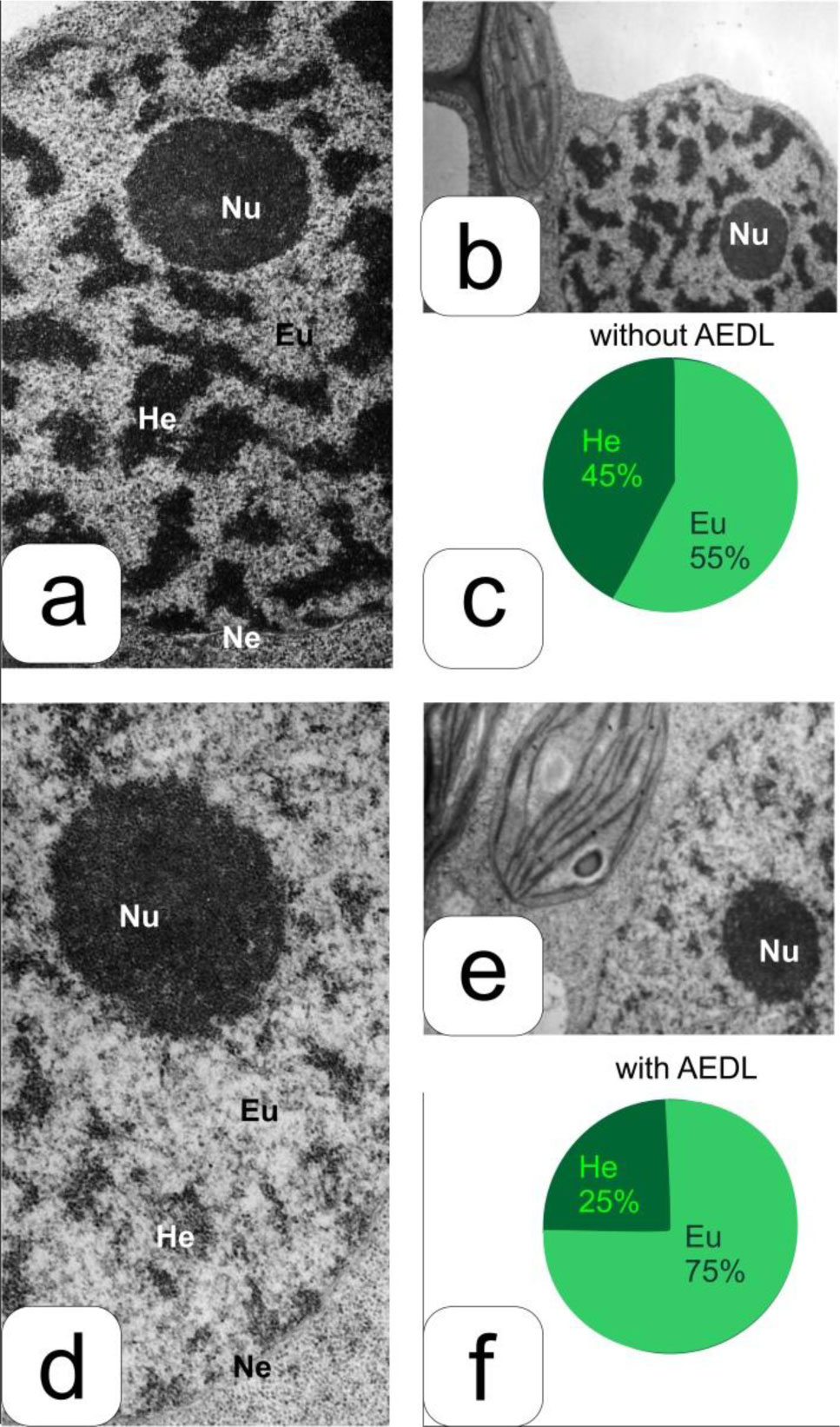
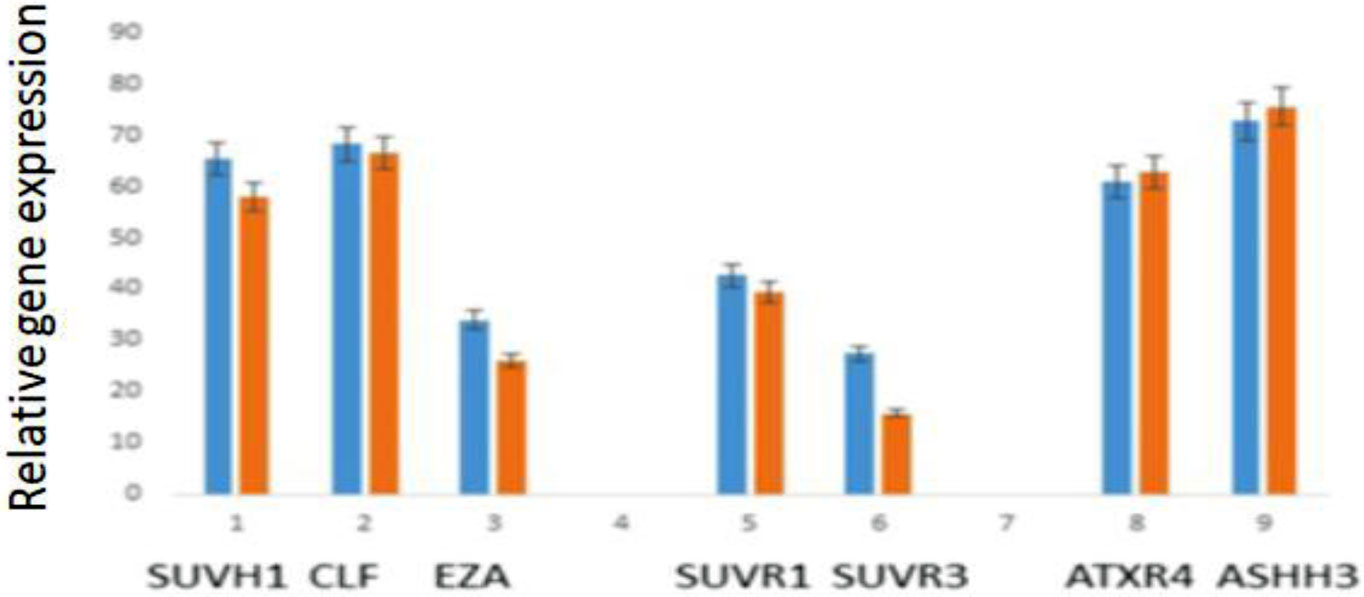
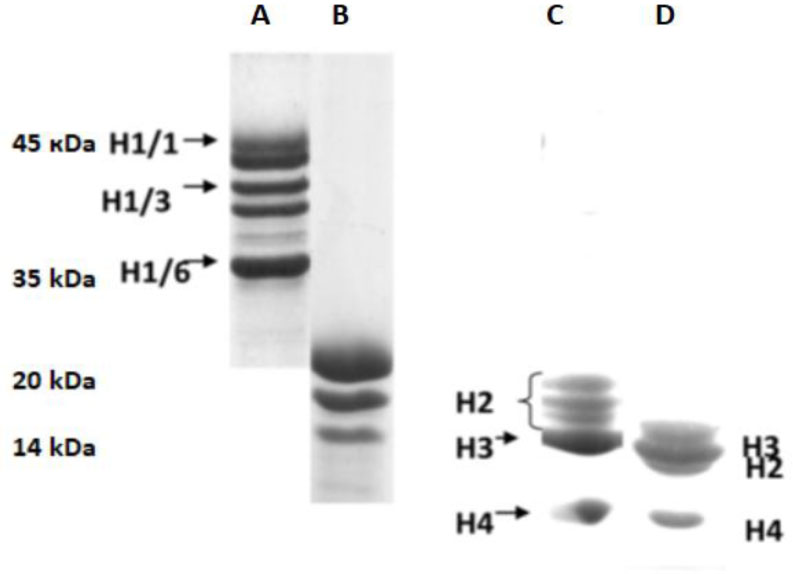
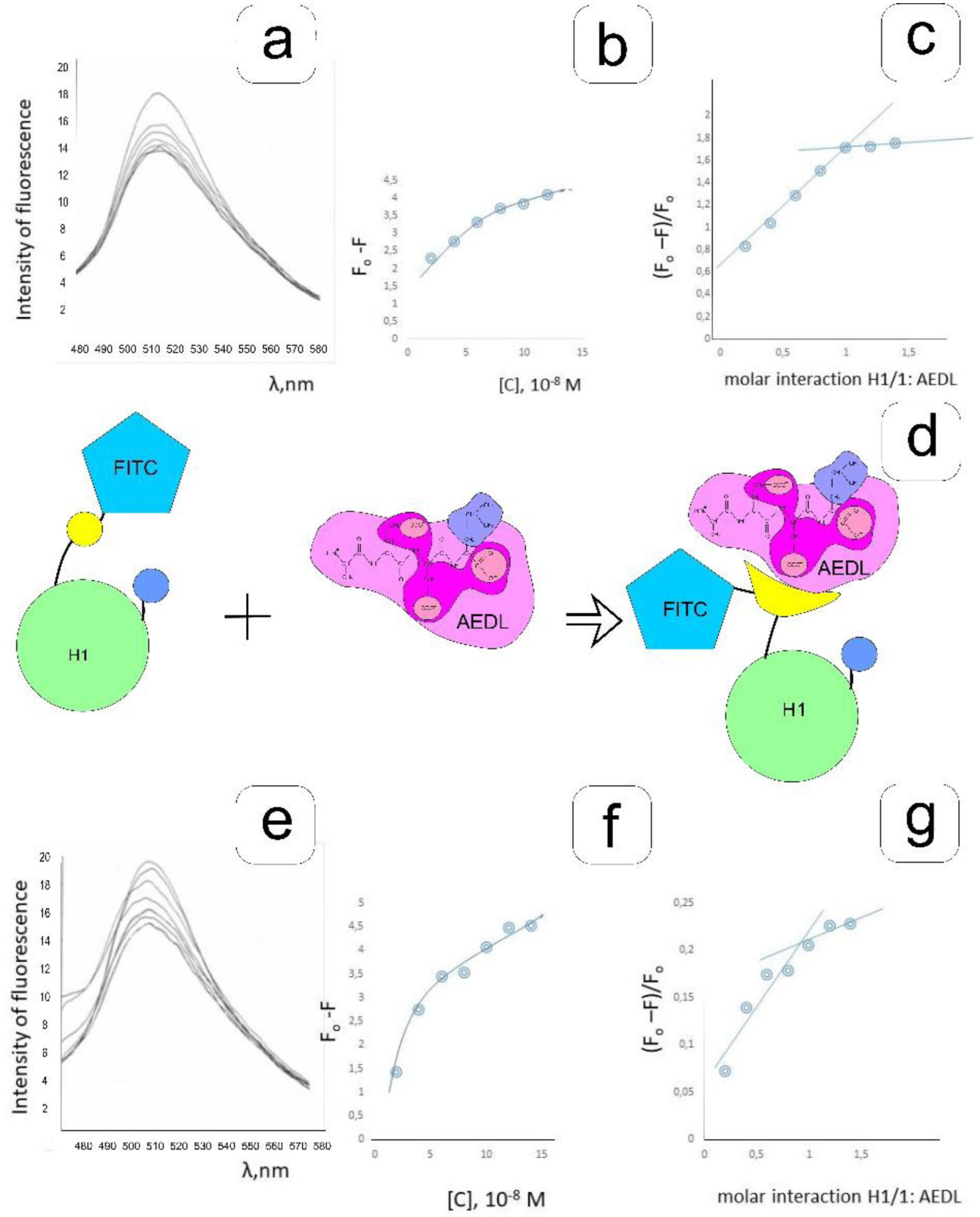
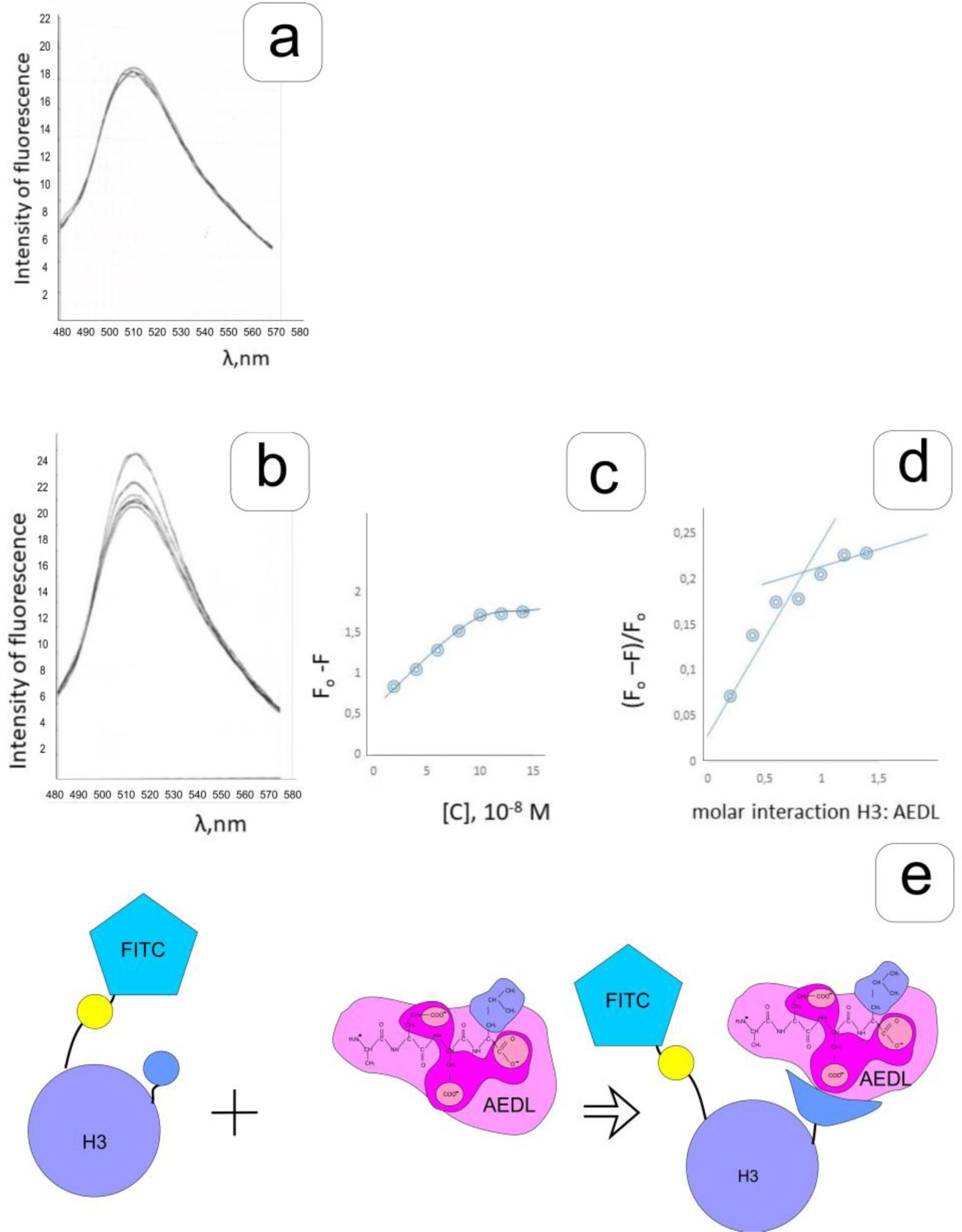
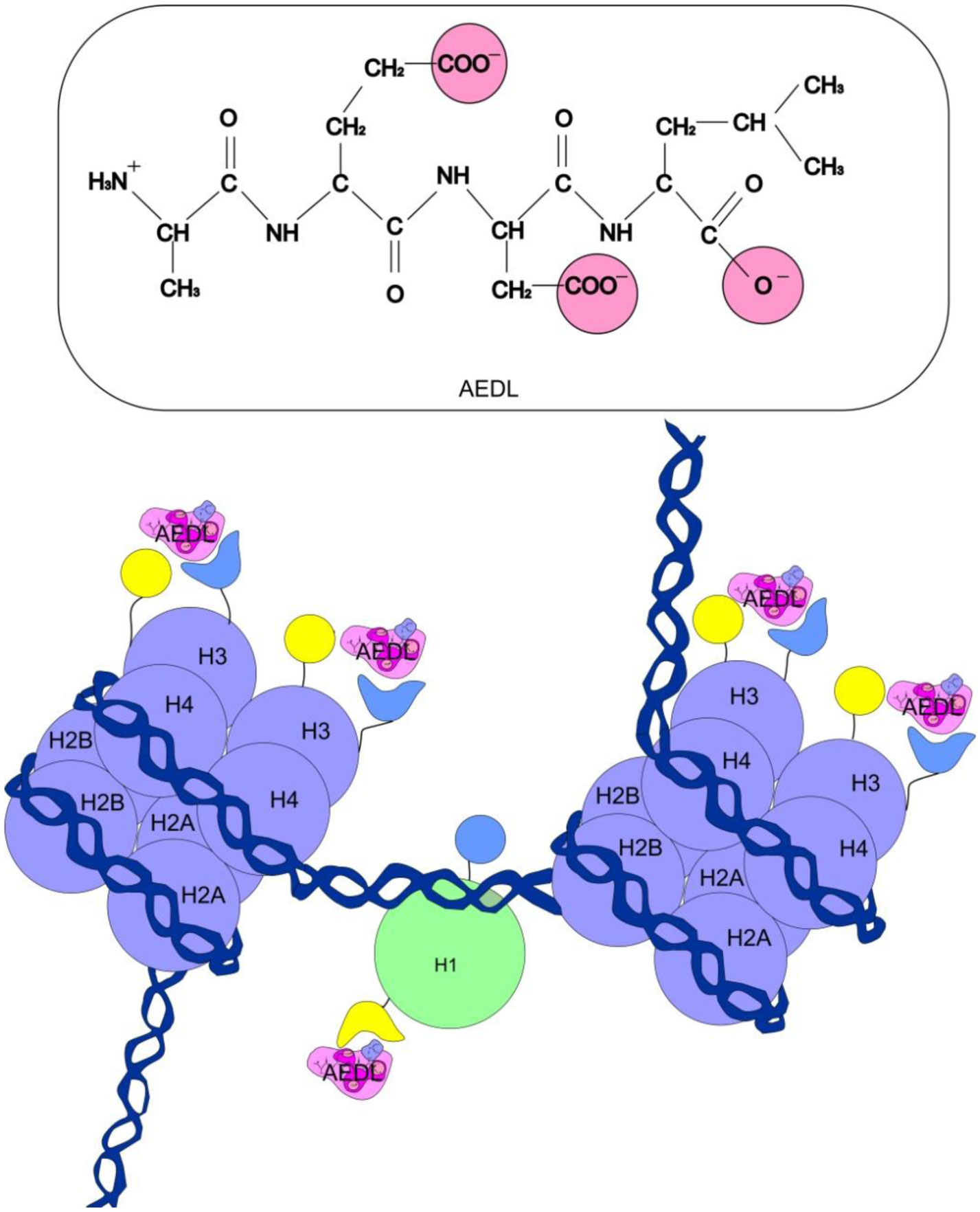
Eukaryotic DNA is tightly packed into chromatin, a DNA–protein structure that exists as transcriptionally permissive euchromatin or repressive heterochromatin. Post-translational modification of histones plays a key role in regulating chromatin dynamics. Short peptides derived from various sources are known to function as epigenetic modulators; however, their mechanisms of action are poorly understood. We addressed this issued by investigating the effect of peptide AEDL on chromatin structure in tobacco (Nicotiana tabacum L.), a commercially important plant species. The chromatin of tobacco interphase cells is characterized by the presence of zones of transcriptionally active domains and particular domains of condensed chromatin of cells that partially coincide with heterochromatin zones. Chromatin decondensation and the formation of euchromatin, accompanied by the activation of genes expression activity, are a determining factor in responses to stressful effects. Our results show that plants grown in the presence of 10−7 M peptide AEDL transformed condensed chromatin domains from 45% in control cells to 25%. Histone modifications, which constitute the so-called histone code, play a decisive role in the control of chromatin structure. Fluorescence quenching experiments using fluorescein isothiocyanate-labeled histones revealed that the linker histone H1 and complexes of core H3 and H1 histones with DNA bound to peptide AEDL in a 1: 1 molar ratio. The peptide was found to bind to the N-terminal lysine residue of H1 and the lysine residue at position 36 of the H3 C terminus. These interactions of histones H1 and H3 with AEDL peptide loosened the tightly packed chromatin structure, getting transcriptionally active euchromatin. Our findings provide novel insight into the mechanism of gene regulation by short peptides and have implications for breeding more resistant or productive varieties of tobacco and other crops.
Citation: Larisa I. Fedoreyeva, Boris F. Vanyushin, Ekaterina N. Baranova. Peptide AEDL alters chromatin conformation via histone binding[J]. AIMS Biophysics, 2020, 7(1): 1-16. doi: 10.3934/biophy.2020001
Eukaryotic DNA is tightly packed into chromatin, a DNA–protein structure that exists as transcriptionally permissive euchromatin or repressive heterochromatin. Post-translational modification of histones plays a key role in regulating chromatin dynamics. Short peptides derived from various sources are known to function as epigenetic modulators; however, their mechanisms of action are poorly understood. We addressed this issued by investigating the effect of peptide AEDL on chromatin structure in tobacco (Nicotiana tabacum L.), a commercially important plant species. The chromatin of tobacco interphase cells is characterized by the presence of zones of transcriptionally active domains and particular domains of condensed chromatin of cells that partially coincide with heterochromatin zones. Chromatin decondensation and the formation of euchromatin, accompanied by the activation of genes expression activity, are a determining factor in responses to stressful effects. Our results show that plants grown in the presence of 10−7 M peptide AEDL transformed condensed chromatin domains from 45% in control cells to 25%. Histone modifications, which constitute the so-called histone code, play a decisive role in the control of chromatin structure. Fluorescence quenching experiments using fluorescein isothiocyanate-labeled histones revealed that the linker histone H1 and complexes of core H3 and H1 histones with DNA bound to peptide AEDL in a 1: 1 molar ratio. The peptide was found to bind to the N-terminal lysine residue of H1 and the lysine residue at position 36 of the H3 C terminus. These interactions of histones H1 and H3 with AEDL peptide loosened the tightly packed chromatin structure, getting transcriptionally active euchromatin. Our findings provide novel insight into the mechanism of gene regulation by short peptides and have implications for breeding more resistant or productive varieties of tobacco and other crops.

| [1] |
Chen DH, Huang Y, Jiang C, et al. (2018) Chromatin-based regulation of plant root development. Front Plant Sci 9: 1509. doi: 10.3389/fpls.2018.01509

|
| [2] |
Luger K, Dechassa ML, Tremethick DJ (2012) New insights into nucleosome and chromatin structure: an ordered state or a disordered affair? Nat Rev Mol Cell Bio 13: 436-447. doi: 10.1038/nrm3382
|
| [3] |
Turner BM (2002) Cellular memory and the histone code. Cell 111: 285-291. doi: 10.1016/S0092-8674(02)01080-2
|
| [4] |
Bannister AJ, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21: 381-395. doi: 10.1038/cr.2011.22
|
| [5] |
Wang N, Liu C (2019) Implications of liquid-liquid phase separation in plant chromatin organization and transcriptional control. Curr Opin Genet Dev 55: 59-65. doi: 10.1016/j.gde.2019.06.003
|
| [6] |
Minard ME, Jain AK, Barton MC (2009) Analysis of epigenetic alterations to chromatin during development. Genesis 47: 559-572. doi: 10.1002/dvg.20534
|
| [7] |
Fischle W, Wang Y, Allis CD (2003) Histone and chromatin cross-talk. Curr Opin Cell Biol 15: 172-183. doi: 10.1016/S0955-0674(03)00013-9
|
| [8] |
Zhang K, Sridhar VV, Zhu J, et al. (2007) Distinctive core histone post-translational modification patterns in Arabidopsis thaliana. Plos One 2: 1210. doi: 10.1371/journal.pone.0001210
|
| [9] |
Lippman Z, Gendrel AV, Black M, et al. (2004) Role of transposable elements in heterochromatin and epigenetic control. Nature 430: 471-476. doi: 10.1038/nature02651
|
| [10] |
Zhang X (2008) The epigenetic landscape of plants. Science 320: 489-492. doi: 10.1126/science.1153996
|
| [11] |
Luger K, Mäder AW, Richmond RK, et al. (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389: 251-260. doi: 10.1038/38444
|
| [12] |
Kalashnikova AA, Porter-Goff ME, Muthurajan UM, et al. (2013) The role of the nucleosome acidic patch in modulating higher order chromatin structure. J R Soc Interface 10: 20121022. doi: 10.1098/rsif.2012.1022
|
| [13] |
McGinty RK, Tan S (2015) Nucleosome structure and function. Chem Rev 115: 2255-2273. doi: 10.1021/cr500373h
|
| [14] |
Grossniklaus U, Vielle-Calzada JP, Hoeppner MA, et al. (1998) Maternal control of embryogenesis by MEDEA, a polycomb group gene in Arabidopsis. Science 280: 446-450. doi: 10.1126/science.280.5362.446
|
| [15] |
Alvarez-Venegas R, Pien S, Sadder M, et al. (2003) ATX-1 an Arabidopsis homolog of trithorax, activates flower homeotic genes. Curr Biol 13: 627-637. doi: 10.1016/S0960-9822(03)00243-4
|
| [16] |
Zhao Z, Yu Y, Meyer D, et al. (2005) Prevention of early flowering by expression of FLOWERING LOCUS C requires methylation of histone H3 K36. Nat Cell Biol 7: 1256-1260. doi: 10.1038/ncb1329
|
| [17] |
Fang Q, Chen P, Wang M, et al. (2016) Human cytomegavirus IE1 protein alters the higher order chromatin structure by targeting the acidic patch of the nucleosome. eLife 5: e11911. doi: 10.7554/eLife.11911
|
| [18] |
Tavormina P, De Coninck B, Nikonorova N, et al. (2015) The plant peptidome: an expanding repertoire of structural features and biological functions. Plant Cell 27: 2095-2118. doi: 10.1105/tpc.15.00440
|
| [19] |
Khavinson V, Popovich I (2017) Short peptides regulate gene expression, protein synthesis and enhance life span. Anti-aging Drugs 496-513. doi: 10.1039/9781782626602-00496
|
| [20] |
Fedoreyeva LI, Dilovarova TA, Ashapkin VV, et al. (2017) Short exogenous peptides regulate expression of CLE, KNOX1 and GRF family genes in Nicotiana tabacum. Biochemistry (Moscow) 82: 521-528. doi: 10.1134/S0006297917040149
|
| [21] |
Khavinson VK, Lezhava TA, Malinin VV (2004) Effects of short peptides on lymphocyte chromatin in senile subjects. B Exp Biol Med 137: 78-81. doi: 10.1023/B:BEBM.0000024393.40560.05
|
| [22] |
Hieb AR, D'Arcy S, Kramer MA, et al. (2012) Fluorescence strategies for high-throughput quantification of protein interactions. Nucleic Acids Res 40: e33. doi: 10.1093/nar/gkr1045
|
| [23] |
Castell JV, Pestaña A, Castro R, et al. (1978) Fluorometric assays in the study of nucleic acid-protein interactions: II. The use of fluorescamine as a reagent for proteins. Anal Biochem 90: 551-560. doi: 10.1016/0003-2697(78)90149-5
|
| [24] |
Lakowicz JR, Weber G (1973) Quenching of fluorescence by oxygen. Probe for structural fluctuations in macromolecules. Biochemistry 12: 4161-4170. doi: 10.1021/bi00745a020
|
| [25] |
Xie MX, Long M, Liu Y, et al. (2006) Characterization of the interaction between human serum albumin and morin. BBA-Gen Subjects 1760: 1184-1191. doi: 10.1016/j.bbagen.2006.03.026
|
| [26] |
Ou HD, Phan S, Deerinck TJ, et al. (2017) ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 357: eaag0025. doi: 10.1126/science.aag0025
|
| [27] | Thorstensen T, Grini PE, Aalen RB (2011) SET domain proteins in plant development. BBA-GENE Regul MechE 1809: 407-420. |
| [28] |
Baumbusch LO, Thorstensen T, Krauss V, et al. (2001) The Arabidopsis thaliana genome contains at least 29 active genes encoding SET domain proteins that can be assigned to four evolutionarily conserved classes. Nucleic Acids Res 29: 4319-4333. doi: 10.1093/nar/29.21.4319
|
| [29] |
Qian C, Zhou MM (2006) SET domain protein lysine methyltransferases: Structure, specificity and catalysis. Cell Mol Life Sci 63: 2755-2763. doi: 10.1007/s00018-006-6274-5
|
| [30] |
Tschiersch B, Hofmann A, Krauss V, et al. (1994) The protein encoded by the Drosophila position-effect variegation suppressor gene Su(var)3–9 combines domains of antagonistic regulators of homeotic gene complexes. EMBO J 13: 3822-3831. doi: 10.1002/j.1460-2075.1994.tb06693.x
|
| [31] |
Rea S, Eisenhaber F, O'Carroll D, et al. (2000) Regulation chromatin structure by site-specific histone H3 methyltransferases. Nature 406: 593-599. doi: 10.1038/35020506
|
| [32] |
Jackson JP, Johnson L, Jasencakova Z, et al. (2004) Dimethylation of histone H3 lysine 9 is a critical mark for DNA methylation and gene silencing in Arabidopsis thaliana. Chromosoma 112: 308-315. doi: 10.1007/s00412-004-0275-7
|
| [33] |
Jasencakova Z, Soppe WJJ, Meister A, et al. (2003) Histone modifications in Arabidopsis–high methylation of H3 lysine 9 is dispensable for constitutive heterochromatin. Plant J 33: 471-480. doi: 10.1046/j.1365-313X.2003.01638.x
|
| [34] |
Neves N, Delgado M, Silva M, et al. (2005) Ribosomal DNA heterochromatin in plants. Cytogenet genome res 109: 104-111. doi: 10.1159/000082388
|
| [35] |
Frapporti A, Pina CM, Arnaiz O, et al. (2019) The Polycomb protein Ezl1 mediates H3K9 and H3K27 methylation to repress transposableelements in Paramecium. Nat Commun 10: 1-15. doi: 10.1038/s41467-019-10648-5
|
| [36] |
Shu J, Chen C, Thapa RK, et al. (2019) Genome-wide occupancy of histone H3K27 methyltransferases CURLY LEAF and SWINGER in Arabidopsis seedlings. Plant Direct 3: e00100. doi: 10.1002/pld3.100
|
| [37] |
Müller-Xing R, Clarenz O, Pokorny L, et al. (2014) Polycomb-group proteins and FLOWERING LOCUS T maintain commitment to flowering in Arabidopsis thaliana. Plant Cell 26: 2457-2471. doi: 10.1105/tpc.114.123323
|
| [38] |
Liu X, Kim YJ, Müller R, et al. (2011) AGAMOUS terminates floral stem cell maintenance in Arabidopsis by directly repressing WUSCHEL through recruitment of Polycomb group proteins. Plant Cell 23: 3654-3670. doi: 10.1105/tpc.111.091538
|
| [39] |
Aichinger E, Villar CBR, Di Mambro R, et al. (2011) The CHD3 chromatin remodeler PICKLE and Polycomb group proteins antagonistically regulate meristem activity in the Arabidopsis root. Plant Cell 23: 1047-1060. doi: 10.1105/tpc.111.083352
|
| [40] |
Ho JWK, Jung YL, Liu T, et al. (2014) Comparative analysis of metazoan chromatin organization. Nature 512: 449-452. doi: 10.1038/nature13415
|
| [41] |
Kizer KO, Phatnani HP, Shibata Y, et al. (2005) A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol 25: 3305-3316. doi: 10.1128/MCB.25.8.3305-3316.2005
|
| [42] |
Schmitges FW, Prusty AB, Faty M, et al. (2011) Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell 42: 330-341. doi: 10.1016/j.molcel.2011.03.025
|
| [43] |
Schuettengruber B, Martinez AM, Iovino N, et al. (2011) Trithorax group proteins: Switching genes on and keeping them active. Nat Rev Mol Cell Bio 12: 799-814. doi: 10.1038/nrm3230
|
| [44] |
Napsucialy-Mendivil S, Alvarez-Venegas R, Shishkova S, et al. (2014) Arabidopsis homolog of trithorax1 (ATX1) is required for cell production, patterning, and morphogenesis in root development. J Exp Bot 65: 6373-6384. doi: 10.1093/jxb/eru355
|
| [45] |
GANTT JS, Lenvik TR (1991) Arabidopsis thaliana H1 histones. Analysis of two members of a small gene family. Eur J Biochem 202: 1029-1039. doi: 10.1111/j.1432-1033.1991.tb16466.x
|
| [46] |
Bednar J, Horowitz RA, Grigoryev SA, et al. (1998) Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. P Natl Acad Sci 95: 14173-14178. doi: 10.1073/pnas.95.24.14173
|
| [47] |
Happel N, Doenecke D (2009) Histone H1 and its isoforms. Contribution to chromatin structure and function. Gene 431: 1-12. doi: 10.1016/j.gene.2008.11.003
|
| [48] |
Favicchio R, Dragan AI, Kneale GG, et al. (2009) Fluorescence spectroscopy and anisotropy in the analysis of DNA-protein interactions. Methods in Mol Biol 543: 589-611. doi: 10.1007/978-1-60327-015-1_35
|

Figures(7)

Larisa I. Fedoreyeva, Boris F. Vanyushin, Ekaterina N. Baranova. Peptide AEDL alters chromatin conformation via histone binding[J]. AIMS Biophysics, 2020, 7(1): 1-16. doi: 10.3934/biophy.2020001







 DownLoad:
DownLoad: