Citation: Agnès Collet, Julien Tarabeux, Elodie Girard, Catherine Dubois DEnghien, Lisa Golmard, Vivien Deshaies, Alban Lermine, Anthony Laugé, Virginie Moncoutier, Cédrick Lefol, Florence Copigny, Catherine Dehainault, Henrique Tenreiro, Christophe Guy, Khadija Abidallah, Catherine Barbaroux, Etienne Rouleau, Nicolas Servant, Antoine De Pauw, Dominique Stoppa-Lyonnet, Claude Houdayer. Pros and cons of HaloPlex enrichment in cancer predisposition genetic diagnosis[J]. AIMS Genetics, 2015, 2(4): 263-280. doi: 10.3934/genet.2015.4.263

| [1] |
Sie AS, Prins JB, van Zelst-Stams WG, et al. (2015) Patient experiences with gene panels based on exome sequencing in clinical diagnostics: high acceptance and low distress. Clin genet 87: 319-326. doi: 10.1111/cge.12433

|
| [2] |
Tarabeux J, Zeitouni B, Moncoutier V, et al. (2014) Streamlined ion torrent PGM-based diagnostics: BRCA1 and BRCA2 genes as a model. Eur j hum genet 22: 535-541. doi: 10.1038/ejhg.2013.181
|
| [3] |
Domchek SM, Bradbury A, Garber JE, et al. (2013) Multiplex genetic testing for cancer susceptibility: out on the high wire without a net? J clin oncol 31: 1267-1270. doi: 10.1200/JCO.2012.46.9403
|
| [4] |
Easton DF, Pharoah PDP, Antoniou AC, et al. (2015) Gene-Panel Sequencing and the Prediction of Breast-Cancer Risk. N engl j med 372: 2243-2257. doi: 10.1056/NEJMsr1501341
|
| [5] |
Audeh MW, Carmichael J, Penson RT, et al. (2010) Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet 376: 245-251. doi: 10.1016/S0140-6736(10)60893-8
|
| [6] | Guttmacher AE, McGuire AL, Ponder B, et al. (2010) Personalized genomic information: preparing for the future of genetic medicine. Nat rev genet 11: 161-165. |
| [7] |
Houdayer C, Caux-Moncoutier V, Krieger S, et al. (2012) Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum mutat 33: 1228-1238. doi: 10.1002/humu.22101
|
| [8] |
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat meth 9: 357-359. doi: 10.1038/nmeth.1923
|
| [9] |
Li H, Handsaker B, Wysoker A, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078-2079. doi: 10.1093/bioinformatics/btp352
|
| [10] |
Koboldt DC, Zhang Q, Larson DE, et al. (2012) VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome res 22: 568-576. doi: 10.1101/gr.129684.111
|
| [11] |
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucl acids res 38: e164-e164. doi: 10.1093/nar/gkq603
|
| [12] |
Ye K, Schulz MH, Long Q, et al. (2009) Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25: 2865-2871. doi: 10.1093/bioinformatics/btp394
|
| [13] |
Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome biol 11: R106. doi: 10.1186/gb-2010-11-10-r106
|
| [14] |
Mertes F, ElSharawy A, Sauer S, et al. (2011) Targeted enrichment of genomic DNA regions for next-generation sequencing. Brief funct genomics 10: 374-386. doi: 10.1093/bfgp/elr033
|
| [15] | Coonrod EM, Durtschi JD, VanSant WC, et al. (2014) Next-generation sequencing of custom amplicons to improve coverage of HaloPlex multigene panels. Biotechniques 57: 204-207. |
| [16] |
Claes KBM, De Leeneer K (2014) Dealing with pseudogenes in molecular diagnostics in the next-generation sequencing era. Methods mol biol 1167: 303-315. doi: 10.1007/978-1-4939-0835-6_21
|
| [17] |
Gréen A, Gréen H, Rehnberg M, et al. (2015) Assessment of HaloPlex Amplification for Sequence Capture and Massively Parallel Sequencing of Arrhythmogenic Right Ventricular Cardiomyopathy-Associated Genes. J mol diagn 17: 31-42. doi: 10.1016/j.jmoldx.2014.09.006
|
| [18] |
Crobach S, Ruano D, van Eijk R, et al. (2015) Target-enriched next-generation sequencing reveals differences between primary and secondary ovarian tumors in formalin-fixed, paraffin-embedded tissue. J mol diagn 17: 193-200. doi: 10.1016/j.jmoldx.2014.10.006
|
| [19] |
Goecks J, Nekrutenko A, Taylor J (2010) Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome biology 11: R86. doi: 10.1186/gb-2010-11-8-r86
|
| [20] | Blankenberg D, Kuster GV, Coraor N, et al. (2001) Galaxy: A Web-Based Genome Analysis Tool for Experimentalists. In: Current Protocols in Molecular Biology. John Wiley & Sons, Inc.; 2001. Available from: http://onlinelibrary.wiley.com/doi/10.1002/0471142727.mb1910s89/abstract |
| [21] |
Giardine B, Riemer C, Hardison RC, et al. (2005) Galaxy: A platform for interactive large-scale genome analysis. Genome res 15: 1451-1455. doi: 10.1101/gr.4086505
|
| [22] |
O'Rawe J, Jiang T, Sun G, et al. (2013) Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing. Genome med 5: 28. doi: 10.1186/gm432
|
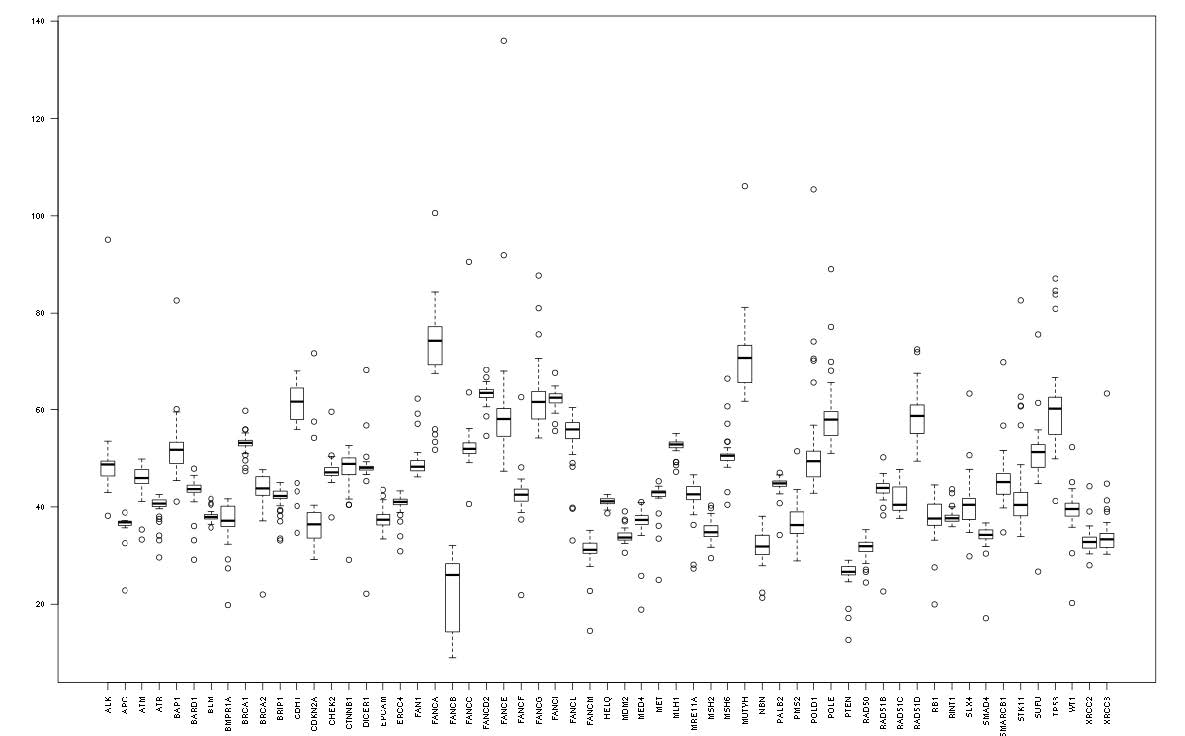
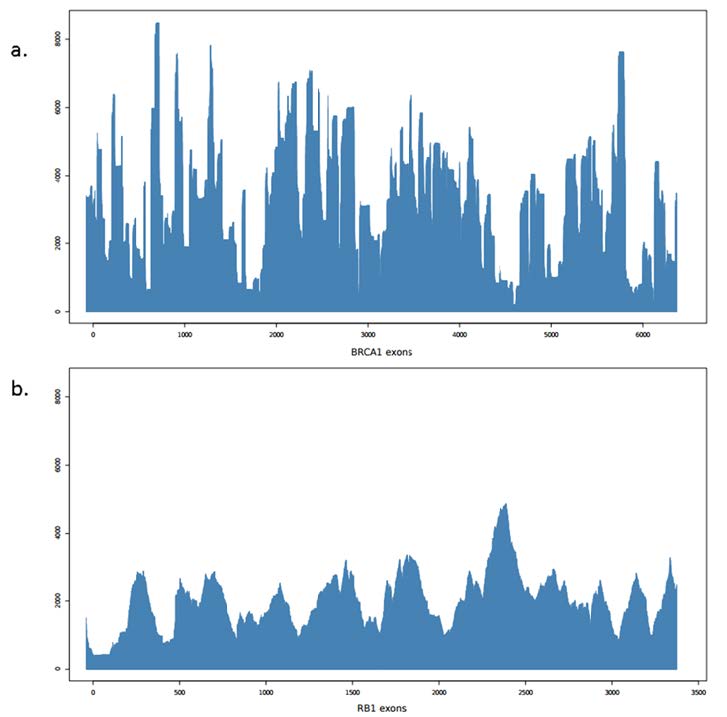
Figures(2) / Tables(4)

Agnès Collet, Julien Tarabeux, Elodie Girard, Catherine Dubois DEnghien, Lisa Golmard, Vivien Deshaies, Alban Lermine, Anthony Laugé, Virginie Moncoutier, Cédrick Lefol, Florence Copigny, Catherine Dehainault, Henrique Tenreiro, Christophe Guy, Khadija Abidallah, Catherine Barbaroux, Etienne Rouleau, Nicolas Servant, Antoine De Pauw, Dominique Stoppa-Lyonnet, Claude Houdayer. Pros and cons of HaloPlex enrichment in cancer predisposition genetic diagnosis[J]. AIMS Genetics, 2015, 2(4): 263-280. doi: 10.3934/genet.2015.4.263







 DownLoad:
DownLoad: